Comparison Chart
Click on a disease to learn more, and to see larger images. Use Symptom Search to look for certain disease features.
| Acronym | Name | SAID group | Gene | Inheritance | Ethnicity | Frequency | Timing of symptoms | Age of onset | Skin cutaneous | Neurologic | Auditory | Ophthalamic | Cardiopulmonary | Abdominal | Lymphatic | Joints bones muscles cartilage | Vasculitis | Amyloidosis | Abnormal labs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Familial Cold Autoinflammatory Syndrome (NLRP3-AID-mild) | Cryopyrin Associated Periodic Syndromes (CAPS) NLRP3-associated Autoinflammatory Diseases (NLRP3-AID) | NLRP3 | Autosomal dominant. Many large family groups spanning generations. Some patients with spontaneous mutations. [1] | Affects all races, but many are of European descent. [1] | 1:1 million, or more. In USA 300+ diagnosed – most cases are from large family groups. [2], [5] Frequency of CAPS in France is 1:360,000. [55] | 12-24 hours, or longer. Onset of fever and flares is often 1-3 hours after exposure to cold or cooling temperatures. [1] | Infancy, but a few present with symptoms later in childhood or adolescence. [1] | Cold-induced, urticaria-like rash with increased neutrophils at the eccrine coils. [4] Almost daily rash that increases with flares. [1] Ice cube test negative. A few with apthous ulcers. | Some have headaches and fatigue with fever after cold exposure. It is unknown if there are any other effects on the central nervous system (CNS) at this time. [1] | Some patients have mild hearing loss – not currently known if it‘s from CAPS inflammation. [1] | Conjunctivitis (non-infectious) during flares. [1] | Not noted. [1] | Uncommon. [1] | Not noted. [1] | Arthralgia, stiffness and swelling with flares. [1] | Not noted. [1] | Elevated serum amyloid (SAA). Secondary amyloidosis in some patients. [1], [9] | High: ESR, CRP, SAA. Leukocytosis with flares. [1] | |
| Muckle-Wells Syndrome (NLRP3-AID-moderate) | Cryopyrin Associated Periodic Syndromes (CAPS) NLRP3-associated Autoinflammatory Disease (NLRP3-AID) | NLRP3 | Autosomal dominant. Spontaneous mutations, and some family groups with MWS spanning generations. [1] | Affects all races, but many are of European descent. [1] | 1:1 million, but it may be more frequent. Some large family groups. [5] Frequency of CAPS in France is 1:360,000. [55] | Often lasts 2-3 days. Random onset–flares of fever and symptoms are often triggered by cold or cooling temperature. [1] | Infancy, but a few present with symptoms later in childhood or adolescence. [1] | Urticaria-like rash with increased neutrophils at the eccrine coils. Rash and flares can be cold-induced, or from unknown triggers. [4] Most have a daily rash that increases with flares. [1] A few have apthous ulcers. Negative ice cube test. | Some have headaches, fatigue with fever and flares. It is uncommon to have many other central nervous system symptoms. [1] A few patients have a MWS/NOMID crossover of symptoms. | Many have increased sensorineural hearing loss, starting in adolescence. [1] | Conjunctivitis (non-infectious) during flares,[1] or corneal haze. [26] MWS/NOMID crossover patients may have more eye involvement. | Rare. [1] | Some have abdominal pain with flares or other gastrointestinal issues. [1] | Rarely noted. [1] | Arthralgia, recurrent arthritis, stiffness and swelling with flares. [1] | Not noted. [1] | Elevated serum amyloid (SAA). >25 % with secondary amyloidosis. [1], [9] | High: ESR, CRP, SAA. Leukocytosis with flares. [1] | |
| Neonatal Onset Multisystem Autoinflammatory Disease - aka Chronic Infantile Neurological Cutaneous Articular Syndrome (NLRP3-AID-severe) | Cryopyrin Associated Periodic Syndromes (CAPS) NLRP3-associated Autoinflammatory Diseases (NLRP3-AID) | NLRP3 | Autosomal dominant. Most cases are due to spontaneous mutations. Very few familial cases. [1] | Any, present in all races. [1] | Estimated frequency 1:1 million, mostly due to spontaneous genetic mutations. [5] | Continuous, with increased symptoms and fever during flares. [1] Chronic inflammation noted between flares. | Neonatal/early infancy. Rash, symptoms, and abnormal labs are often present at birth. [1], [6] | Ever-present, urticaria-like rash with increased neutrophils at the eccrine coils. Rash increases with flares. Some patients have cold-induced flares in addition to constant symptoms. [1] [4] A few with apthous ulcers. Negative ice cube test. | Headaches, fever, fatigue, chronic aseptic meningitis, and elevated or high intracranial pressure (ICP). Papilledema is common. Many have mental delay and/or cognitive delay, impairments, or intellectual disability. A few have seizures. Strokes are rare. [6] | Many have increased sensorineural hearing loss, starting in infancy/early childhood. [1], [6] | Papilledema, uveitis, iritis, conjunctivitis. Some with retinal scarring, corneal haze or vision loss. [6], [26] | Some may have a pericardial effusion, or pericarditis. [1] | Some patients have hepatomegaly, splenomegaly, or hepatosplenomegaly. Nausea, vomiting and abdominal pain with flares, or with elevated intracranial pressure (ICP). [6] | Splenomegaly. Many have generalized lymphadenopathy. [1] | Joint pain, knee valgus or varus, limb length differences are common. Some have frontal bossing of the forehead, saddleback nose, contractures, and/or have clubbing of the fingers. [1] Short stature, growth delays, failure to thrive, arthritis, and osteopenia are often noted.[1],[26] <50% of patients knees or joints have bony overgrowth (usually on the patella) with chondrocytes that are not well differentiated, plus abnormal enchondral bone formation (no inflammatory cells), along with the premature fusion of physis. [79]. | Vasculitis rarely develops. [1] | Elevated SAA. Secondary amyloidosis in <2% pts. [1], [6] | Chronically high: ESR, CRP, SAA, anemia, granulocytic leukocytosis. Many patients have elevated IgG, IgA and IgM [1], [6] | |
| Schnitzler Syndrome | Schnitzler Syndrome | Currently unknown. | Unknown. | Affects all races, but most cases are in Europe. [13] More men than women are affected. | Unknown. Over 150 known cases, mostly in Europe. [13] | 12-36 hours. Rash is present first. Intermittent fevers often occur separately from the rash. [13] | Most cases start in middle age, at over 35-50 years old. The youngest patient was 13 yrs old. Symptoms start with the rash. [13] | Maculopapular rash and plaques (sometimes itchy) on the chest and limbs. Dermis has neutrophillic infiltrate. Dermographism. [13] | Intermittent fevers can rise >40°C. Chills are uncommon. Fatigue and headaches are common with fevers. Temperature changes (weather), stress and/or exercise can trigger flares. [13] | Uncommon. [13] | Not noted. [13] | Not noted. [13] | GI symptoms are uncommon. Hepatomegaly, splenomegaly or hepatosplenomegaly is common. [13] | <20% with lymphoma, IgM myeloma, or Waldenströms macroglobulinemia (aka lymphoplasmacytic lymphoma). >45% with lymphadenopathy. Splenomegaly. [13] | 80% have muscle, bone and/or joint pain, or arthritis. Bone pain is most common in the iliac and tibia. <40% have bone lesions. Some with osteocondensation and sclerotic bone marrow involvement in the legs. [13] | Vasculitis noted in 20% of patients. [13] | A few patients have developed secondary amyloidosis. [13] | Monoclonal IgM and/or IgG gammopathy. High: ESR, CRP. Leukocytosis. Complement normal to elevated. 50% with inflammatory anemia. [13] | |
| Familial Mediterranean Fever | Pyrin-associated Autoinflammatory Diseases (PAAD) | MEFV | Autosomal recessive in the majority of patients. Some cases have gene-dosage-dependent autosomal dominant inheritance. [10] | Turk, Armenian, Arab, Sephardic Jew, Italian. [1] FMF is the most common inherited periodic fever syndrome. | In specific ethnic groups, the carrier frequency of MEFV variants is up to 1:5 people. [1] | 12-72 hours. [1] [9] Recurrent fever and flares can occur weekly, or only a few times a year. | Infancy, to under 20 years of age for the onset of the first symptoms. [9] Adult-onset is uncommon, but can occur. | Erysipeloid (erysipelas-like) erythema on the ankle–foot–below knee region that lasts 2-3 days during flares of symptoms. [1] | Fevers. Acute aseptic meningitis is rare and can occur during flares, but is never chronic. [1] Other neurological involvement is very rarely seen in FMF. | Uncommon–not believed to be caused by a FMF disorder. [1] | Very rare to uncommon. [1] | 45% have pleuritis, painful respiration with flares. Some with pericarditis. [1] | Sterile peritonitis, pain, and/or constipation with flares. Splenomegaly. [1] Some cases of inflammation causing appendicitis symptoms, but the appendix is inflamed, not infected. | Splenomegaly is common. Some have lymphadenopathy. [1] | Mono or polyarthritis, oligoarthritis and clubbing are common. Ankle arthralgias are common. Severe arthritis of the hip or ankle is rare. [1] | Henoch-Schönlein purpura (HSP), polyarteritis nodosa (PAN). [1] | Secondary Amyloidosis is common. >50% in untreated patients; it depends on genotype. [9] | High: ESR, CRP, SAA between flares. Fibrinogen, leukocytosis present with flares. [1] M694V and some with V726A mutations have higher risk for elevated IgD, and higher risk of more notable FMF symptoms, especially arthritis. [127] Elevated serum IgD levels 10% to 13% of patients with FMF (and TRAPS) [128] | |
| Tumor Necrosis Factor (TNF) - Associated Periodic Syndrome (aka Familial Hibernian Fever) | TNF-associated Autoinflammatory Diseases, Protein Folding | TNFRSF1A | Autosomal dominant. Spontaneous mutations, with some familial groups. [1] | Affects all races. Second most common inherited SAID (after FMF). [1] | Unknown. TRAPS affects 0.01:10,000 people in the European Union. [51] >1000 patients worldwide. [52] | Days to weeks. An average flare lasts around three weeks. [1] [9] | Most first attacks occur by 3 years of age, and almost all begin by 20 years of age. A few have symptoms start later in life. [9] | Migrating rash with deep pain under the areas with the rash. Severe pain follows the rash path from the trunk outwards to the limbs. [9] | Fevers lasting >3 days at over 38°C with flares. Some have headaches with flares of symptoms. [1] [9] | Uncommon–not believed to be caused by TRAPS. [1] | Conjunctivitis, and periorbital edema during flares. [1] [9] | Common, including pleurisy. [1] | Abdominal pain, peritonitis, diarrhea, and constipation with flares. Splenomegaly. [1] | Splenomegaly is common; some have lymphadeopathy. [1] | Intermittent or chronic arthritis in the large joints with muscle pain and swelling is common. [1] | Henoch-Schönlein purpura (HSP), lymphocytic vasculitis. [1] | 10-20% occurrence. Higher risk with a cysteine mutation. [9] | High: ESR, CRP, SAA. Polymorphonuclear neutrophils (PMNs), polyclonalgammopathy, leukocytosis. [1] Elevated serum IgD levels10% to 13% of patients with TRAPS (elevated in some with other autoinflammatory diseases too, such as HIDS, MA, FMF, PFAPA) [128] | |
| TNFRSF11A-associated hereditary fever disease (TRAPS11) | TNF-associated autoinflammatory diseases, Protein Folding | TNFRSF11A | Autosomal dominant. | Not noted. | Unknown-rare | Flares every month that last from 8 days to 3-5 weeks. Some note triggers for flares, such as cold temperatures or pregnancy. [101] | First flares of symptoms in infancy or childhood. Symptoms are often similar to TRAPS. [101] | Macular rash, or erythema nodosum noted in some patients. One case with recurrent pharyngitis. [101] | Recurrent fevers with flares, plus headaches, asthenia and pain. One patient with cold-triggered or pregnancy triggered flares. [101] | Not noted. [101] | Anterior uveitis. [101] | Occasional thoracic pain and one patient with a hacking cough during flares. [101] | Abdominal pain, nausea and/or constipation. Mesenteric adenitis and abdominal pain can mimic appendicitis. [101] | Lymphadenopathy, mesenteric adenitis [101] | Mylagia, arthrlagia, and arthritis (can be severe). One patient with a stress fracture and dental problems, possibly due to amelogenesis imperfecta. [101] In some other types of TNFRSF11A mutations that are NOT associated with periodic fevers and flares, familial expansile osteolysis, osteopetrosis, and Paget disease of bone have been noted, but have NOT been found in the group of patients with the periodic fever type of presentation of symptoms. [90] [101]. | Not noted. [90] [101] | Unknown. Not noted. [90] [101] | High during flares, or sometimes between flares: CRP, ESR, hypergammaglobulinema. One patient with fluctuating in ANA titers but no autoimmunity noted. [101] | |
| Mevalonate Kinase Deficiency-mild/moderate, Hyperimmunoglobulinemia D with Periodic Fever Syndrome | Mevalonate Kinase Deficiency (MKD) | MVK | Autosomal recessive. Some cases with only one mutation found. [33] | Mostly of Dutch descent, or Northern European. [1] | Unknown, but very rare. >200-300 known patients worldwide, (>300, when suspected cases are also included.) [12] | 3-7 days duration. Recurrent bouts of fever and flares every 2-12 weeks. [1] [9] Some flares occur after vaccines. [9] | >90% present with symptoms in infancy. [9] | Diffuse maculopapular rash. Polymorphous rashes. Some with petechiae or purpura present. [1], [9] 50% with apthous ulcers or genital ulcers. [130] Porokeratosis of Mibelli in one patient, and disseminated superficial actinic porokeratosis (DSAP) in a few cases of patients of Asian ancestry. [136] | Headaches and fevers with flares of symptoms are common. [1] [9] More severe neurological symptoms are rarely present in HIDS. [9] | Uncommon – not believed to be caused by HIDS. [1] [9] | Uncommon. [9] Some with conjunctivitis. [134] and there are a few patients with retinitis pigmentosa (RP). [136] | Rare. [1] Some patients have developed severe respiratory infections. Higher risk for issues with S. pneumoniae infections. [78] | Extreme pain, vomiting and diarrhea with flares. [1], [9] A few cases with colitis, including early-onset, and sometimes severe colitis in the neonatal period, with bloody diarrhea. [136]. Some with hepatomegaly, splenomegaly, hepatosplenomegaly, or other gastrointestinal issues. [78] | Cervical lymphadenopathy with flares. [1] A few with splenomegaly. [78] A few patients have had macrophage activation syndrome (MAS.) [136] | Arthralgias are common, symmetric polyarthritis is frequently noted. [1] | Cutaneous vasculitis is common. Henoch-Schönlein purpura (HSP) is rare. [1] | <5-10% – uncommon. [9] | High: ESR, CRP, SAA with flares. Mevalonate aciduria noted during flares. [1] High IgD with IgA in 80% patients. IgD may be normal in infants and young children with HIDS. Note that there are other conditions where IgD may sometimes be elevated, (see also FMF, TRAPS, PFAPA on our chart) so this is not considered the most accurate diagnostic lab for MVK diseases. | |
| Mevalonate Aciduria (MA), Mevalonate Kinase Deficiency-severe | Mevalonate Kinase Deficiency (MKD) | MVK | Autosomal recessive. | Mostly of Dutch descent, or Northern European. [1] | Unknown, but very rare. <100 known patients worldwide. [11] | 4-5 days. Recurrent flares with fevers every 2-3 weeks. Patients have chronic inflammation noted between flares. [11] | Most present with symptoms at birth, or in early infancy. Most have facial features noted at birth. [11] | Diffuse maculopapular or morbilliform rash. Some with petechiae or purpura present. A few with apthous ulcers. [1], [9], [11] | Fevers with flares. Microcephaly, dolichocephaly, intellectual disability, cognitive and/or developmental delays, cerebellar ataxia, cerebellar atrophy and seizures (epilepsy) often develop over time. [11] Patients with Mevalonate Kinase Deficiencies can have flares triggered by vaccines. | Uncommon–not believed to be caused by MA. [1] [9] [11] | Uveitis, central cataracts, blue sclerae and tapetoretinal degeneration are often present, even in less severe cases. [11] | Rare. [1] [11] | Hepatomegaly, splenomegaly or hepatosplenomegaly. Cholestatic liver disease. Abdominal pain, vomiting and diarrhea with flares. [1] [9] [11] One case with hepatic fibrosis. [135] | Splenomegaly, and/or lymphadenopathy are common. [1], [11] | Congenital defects are often noted: microcephaly, dolichocephaly, wide irregular fontanels, low set and posteriorly rotated ears, downslanted palpebral fissures. Hypotonia, myopathy, arthralgia, arthritis and failure to thrive are common. [11] | Not noted. [11] | Not noted-unknown. [9] [11] | Anemia, leukocytosis, thrombocytopenia. High: ESR, CRP, SAA, CK, IgD, IgA, IgE; chronically high Mevalonate aciduria. [1] [11] | |
| Deficiency of the Interleukin-1ß (IL-1ß) Receptor Antagonist (DIRA) (aka Osteomyelitis, Sterile Multifocal with Periostitis Pustulosis) | Inflammatory Bone Diseases | IL1RN | Autosomal recessive. | Carriers in 0.2% population of Newfoundland and 1.3% in Puerto Rico. Some North American, Dutch, Brazilian and Lebanese patients. [16] | Unknown, but very rare. In some parts of Aricibo, Puerto Rico there are more DIRA carriers, so DIRA may occur in 1:6300 people there. [16] | Continuous inflammation from birth/fetal development. Untreated DIRA can lead to death in infancy or early childhood. [16] | Most have symptoms at birth, or as a neonate, such as: pustular rash, bone pain, swollen joints, and apthous ulcers. [16] | Epidermal neutrophilic pustules at hair follicles. Aphthous ulcers, stomatitis, pathergy, hyperkeratosis, acanthosis; high neutrophil infiltrate of the dermis. [16] [26] | High fevers are NOT common, or noted in the neonatal period. Neurological complications are not common. A few cases of cerebral vasculitis noted. [16] [26] | Not noted. [15] [16] | Eye issues are rare. Non-infectious conjunctivitis may be caused by DIRA. [15] [16] | Some with respiratory distress. One known case of pulmonary hemosiderosis with progressive interstitial fibrosis. [15] [16] [17] | DIRA patients rarely have gastrointestinal issues. Failure to thrive is common. Hepatomegaly, splenomegaly or hepatosplenomegaly is common. Risk of organ failure if DIRA is untreated. [16] | Splenomegaly is common.[16] | Joint swelling and severe bone pain. Bone biopsies show no infection. Common: Balloon-like widening of the anterior rib ends, periosteal elevation along multiple long bones, multifocal osteolytic lesions. Other bones may be affected. [16] | A few with localized or cerebral vasculitis. [16] | Not noted. [15] [16] [17] | High: ESR, CRP, leukocytosis, chronic anemia. [16] [56] | |
| Majeed Syndrome – aka Chronic Recurrent Multifocal Osteomyelitis, Congenital Dyserythropoietic Anemia and Neutrophilic Dermatosis Syndrome | Inflammatory Bone Diseases | LPIN2 | Autosomal recessive. Two LPIN2 mutations are required to cause symptoms of Majeed. | Currently, the only documented cases of Majeed are of Middle Eastern ancestry. [18] | Unknown, but very rare. Very few documented cases at this time. [18] [53] | Flares last for a few days, with 1-4 exacerbations a month of high fevers, severe pain, and joint swelling. [18] [53] | Most present with symptoms in infancy to early childhood, starting between 3 weeks to 2 years of age. [18] | Most patients have inflammatory dermatosis, Sweet’s syndrome, pustular skin lesions, psoriasis. Intra-epidermal neutrophils. [18] [53] | High fevers last for a few days with flares and severe pain. Other neurological symptoms are not noted. Growth delays in height, and chronic pain are common. [18] [53] | Not noted. [18] [53] | Not noted. [18] [53] | Not noted. [18] [53] | Hepatomegaly and cholestatic jaundice in the neonatal period, but it is transient. [18] [53] | Neonates: hepatomegaly and neutropenia; anemia is common and can be severe. [18] | Periarticular tender soft tissue swelling. Bone pain. Bone biopsy shows no infection. Early-onset Chronic Recurrent Multifocal Osteomyelitis (CRMO). Periarticular tender soft tissue swelling, short stature, delayed bone age, contractures are often noted. [18] | Not noted. [18] [53] | Not noted. [18] [53] | Congenital dyserythropoietic anemia (CDA). High ESR. WBC can be normal, or elevated, with neutropenia in infancy. Cultures are negative. [18] | |
| Chronic Nonbacterial Osteomyelitis (CNO): Chronic Recurrent Multifocal Osteomyelitis (CRMO); and Synovitis, Acne, Pustulosis, Hyperostosis, Osteitis Syndrome (SAPHO) | Inflammatory Bone Diseases | Currently unknown. No genetics tests available (link has OMIM info about genetic research for CRMO.) | Currently unknown. | Affects all races, but the majority of patients have European ancestry. There are more female patients than males. [21] [22] | Unknown, but rare. | At least 6 months with chronic or relapsing symptoms. Often patients suffer for 7-25 years with symptoms. Many bone lesions heal completely. [19] [22] | Mostly affects children – some adult onset. Peak incidence of flares is around 10 years of age. [22] | Some patients have acne, and/or pustulosis on the palms and/or soles of their extremities (palmoplantar pustulosis – often associated with SAPHO). 23% have psoriasis.[19] [22] [54] | Fevers affect a number of patients during flares of CRMO. Other neurological symptoms are not noted. Some with impaired bone growth, or overall impaired growth.[19] [22] [54] | Not noted. [19] [21] [22] [54] | Some cases of uveitis. [19] | Not common–some patients also have ANCA+ vasculitis that can affect the lungs. [18] [54] | Some patients also have inflammatory bowel diseases. [19] | Some cases of ANCA+ vasculitis that can affect the kidneys. [19] | Joint swelling, limp, severe bone pain over affected bones (mostly the long bones). Some have jawbone involvement. 2-18 bone lesions are commonly found. Earlier age of onset along with many bone lesions is associated with more severe disease. Bone biopsy and cultures show no infection. [19] [22] Chronic nonbacterial osteomyelitis (CNO) is considered part of the spectrum of bone diseases with CRMO and SAPHO, but may be more self-limiting, and less chronic than CRMO or SAPHO. [115] | Some with Takayasu arteritis, or ANCA+ vasculitis. [54] | Not noted. [19] [22] [54] | Whole body MRI can reveal multifocal bone lesions. [20] Normal or elevated WBC, ESR, CRP. [19], [22], [54] Radiographs may appear normal at the start of CNO/CRMO, but an MRI will detect lesions. [81] | |
| IL-10 deficiency-associated inflammatory bowel disease, Early-Onset Inflammatory Bowel Disease; also Very Early-Onset Inflammatory Bowel Disease (<2yrs of age). Subclassifications: EO-IBD25 (IL10RB), EO-IBD28 (IL10RA) and EO-IBD with IL-10 deficiency (IL10) | Il-10 deficiency-associated inflammatory bowel disease | IL10RA, IL10RB, IL10 (deficiency) | Autosomal recessive. | Not noted. | Rare but 25% of the cases of VEO-IBD have been found to be caused by IL-10 mutations. [98] | Continuous, with flares of increased symptoms, such as abdominal pain, rectal bleeding and diarrhea. A few rare cases with fevers and arthritis. [98] | Onset in the first few years of life, often in early infancy. Early onset of severe enterocolitis, Crohns or ulcerative colits, perianal fistuas, and failure to thrive or impaired growth. [120] [98] | Recurrent folliculitis, perianal abscesses. [120] [98] Some cases with aphthous ulcers. It is recommended to also look for erythema nodosum or pyoderma gangrenosum or vitiligo, but these are uncommon. [98] | Rare: Fevers. No neurologic deficits noted. [98] | Not noted. [120] [98] | Not noted. [120] [98] | Not noted. [120] [98] | First symptoms in VEO-IBD is blood in the stools (hematochezia). Abdominal pain, diarrhea and diagnostic findings for Crohns, colitis, ulcerative colitis are noted early in childhood. Perianal fistulas and other abnormalities of the lower GI tract due to the disease state are found. Failure to thrive and delayed growth are common. [98] | Not noted. [120] [98] | Some cases with joint pain, sacroiliiac pain and arthritis have been noted, but the typical presentation does not involve the joints. Growth delay, including short stature is often seen. [123]. | Not noted. [120] [98] | Not noted. [120] [98] | CRP, ESR, platelets may be elevated during flares. Low hemoglobin, albumin, anemia are common. [123] Diagnosis requires endoscopy and colonoscopy, and other tests. Hematochezia (blood in the stool). [122] | |
| Deficiency of the Interleukin-36-Receptor Antagonist (DITRA) – aka Generalized Pustular Psoriasis (GPP) | Pyogenic Diseases | IL36RN | Autosomal recessive. | May affect all races. Patients with Caucasian, Spanish, Asian, African, Algerian or Tunisian ancestry. [58] [75] [77] | Unknown but rare. 1% of Sfax, Tunisians are carriers, with a 0.52% chance of having the disease in this population. [58] | Flares last days to weeks. Some with chronic symptoms. Most flare from triggers like: infections, stress, medication changes, during pregnancy or menstruation. [58] | Variable age of onset. Many have symptoms starting in childhood. Some have symptoms beginning in adulthood. [58] | Recurrent, generalized pustular psoriasis and high fevers after erythematous rash. Some with acral pustules and nail damage, or chronic plaques. [58] [61] | Sudden-onset high fever >40°C with chills. Some patients have a headache with the onset of the rash and fever, plus asthenia (muscle weakness) and tachycardia. [58] [61] | Not noted.[58] [61] [62] | Not noted. [58] [61] [62] | Tachycardia. Electrolyte imbalances can occur during fever and onset of pustular rash. Risk for cardiac arrest and septicemia. [58] [61] | Nausea during flares. At risk for a loss of appetite. [58] [60] One infant patient noted with failure to thrive and diarrhea. [63] | Risk for renal and liver impairment and systemic infection with severe flares. [58] [61] | Asthenia (muscle weakness) during fevers and flares. Risk for inflammatory arthritis. [59] [61] | Not noted. [59] [60] | Not noted. [59] [60] | High during flares (in most patients): ESR, CRP, neutrophils, lactate levels. Low: plasma albumin, calcium, zinc. Risk for infections with flares. [59] [60] | |
| CARD14-associated psoriasis, aka Familial Psoriasis (PSORS2), or CARD14-Mediated Pustular Psoriasis | CARD14-associated psoriasis, Pyogenic Diseases | CARD14 | Autosomal dominant. Spontaneous mutations, and some familial groups. [23] | Most with European or Asian ancestry. Known patients in US, EU, Canada (Newfoundland), Haiti, and Taiwan. [23] | Unknown, but rare. | Continuous. Chronic pustular or plaque psoriasis triggered by inflammatory stimuli. Some cases with psoriatic arthritis. [23] [24] | Variable age of onset from infancy or childhood to adulthood with pustular psoriasis. [23] [24] | Generalized pustular psoriasis (that can be severe), and/or plaque psoriasis. Sometimes nails are affected with psoriasis. [23] [24] | Not seen. [23] [24] | Not seen. [23] [24] | Not seen. [23] [24] | Not seen. [23] [24] | Not seen. [23] [24] | Not seen. [23] [24] | Intermittent joint pain, psoriatic arthritis. 30% of affected patients in one European family with PSORS2 also had psoriatic arthritis. [24] | Not seen. [23] [24] | Not seen. [23] [24] | Mildly elevated WBC. CRP and ESR are rarely elevated – and only during flares of symptoms. [56] | |
| Pustular Psoriasis (15) | Pyogenic Diseases | AP1S3 | Autosomal dominant. | Not noted. | Rare | Pustular flares can be triggered by infections, pregnancy, or certain medications. [121] [122] | Varied onset. [121] [122] | Many cases with the Acrodermatitis continua of Hallopeau (ACH) type of pustular psoriasis that affects the tips of the fingers and toes. Others with palmoplantar, acral or generalized pustular psoriasis (GPP). [98] [122] Sterile pustules, scaling, nail dystrophy, and digit tapering. [121] | Not noted. [121] [122] | Not noted. [121] [122] | Not noted. [121] [122] | Not noted. [121] [122] | Not noted. [121] [122] | Not noted. [121] [122] | Fingertips are affected, and the nails. No indication of joint involvement. [121] | Not noted. [121] [122] | Not noted. [121] [122] | Unknown - not stated in the literature. [121] [122] | |
| PSTPIP1-associated Arthritis, pyoderma gangrenosum and acne (PAPA), aka Pyogenic Sterile Arthritis, Pyoderma Gangrenosum, and Acne Syndrome | Pyogenic Diseases | PSTPIP1 | Autosomal dominant. Spontaneous mutations, with some familial groups. [29] [30] | Currently, the only documented cases are from Europe, New Zealand and the USA. [30] | Unknown, but rare. | Early-onset, destructive, recurrent inflammation of the joints, skin and muscle. Flares often occur after mild injury, or injections. [29] | First symptoms of arthritis develop by 1-10 years of age, and skin lesions develop during adolescence. [29] [32] | Pathergy. Pyoderma gangrenosum ulcerative lesions, and/or severe cystic acne. Affected tissues with high neutrophil infiltration. [29] | Fevers can accompany flares of joint inflammation and pain. Other neurological symptoms are not noted. [31] | Not noted. [29] [30] [31] | Not noted. [29] [30] [31] | Not noted. [29] [30] [31] | Some patients also have irritable bowel syndrome. [29] | Not noted. [29] [30] [31] | Episodic inflammatory arthritis, often affecting one joint at a time that doesn’t resolve on it’s own. Intermittent sterile pauciarticular, peripheral erosive arthritis. Joint damage and destruction can often develop from the arthritis. [29] [30] [31] [32] [55] | Not noted. [29] | Not noted. [29] | Cultures of the bone and skin are negative. Purulent synovial fluid is full of neutrophils. High with flares: CRP, ESR, WBC. [29] [30] [32] | |
| Congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD) | Generation of Intracellular Stress; Congenital Sideroblastic Anemias (CSAs) | TRNT1 | Autosomal recessive. | Unknown | Unknown-rare. | Flares every 2-4 weeks lasting 5-7 days. Some have predictable fever and flare patterns, but most have random attacks, ranging from weekly, to every 3-4 weeks. One case had weekly fevers in infancy, that become bi-monthly in childhood. Periodic fevers with vomiting, and diarrhea starting in infancy for most cases. [89] | Most have symptoms present in the neonatal period or prior to 3 months of age. Once case with onset at 18 months. [89] | Rash is uncommon. Some with notable pallor at birth. A few with brittle hair. [89] One case with chronic icthyosis, erythema and/or hypopigmentation of the skin. Biopsy showed perivascular lymphohistiocytic infiltrate and electron microscopy found a few spots of fibrillar amyloid-like material. [89] | Periodic fevers, progressive cognitive delay and developmental delay, speech and comprehension challenges, cerebral atrophy. [89] Seizures, some with cerebellar ataxia. Neurodegeneration, abnormalities in the cerebellum. [89] | Sensorineural hearing loss. [89] | Retinitis pigmentosa. One case of atypical retinitis pigmentosa and variant retinitis punctata albescens. [89] | Dilated cardiomyopathy. [89] Cardiac failure was the leading cause of death in most cases. [89] [91] Immunodeficiency and higher risk of sinusitis, pneumonia/pulmonary infections (not during SIFD flares). [89] | Vomiting and diarrhea with fever and flares. Mild hepatomegaly and splenomegaly (hepatosplenomegaly) Nephrocalcinosis. [89] High risk of death from multi-organ failure. [91] Cyclic vomiting with metabolic acidosis, feeding poorly, gastrointestinal upset, abdominal pain, and other intestinal issues accompanied by fevers. [89] | Immunodeficiency. Lymphadenopathy, splenomegaly. Once case of fatal adrenal hemmorhage. [89] | Hypotonia (generalized or tructal) with increasing severity. Gross motor delay and developmental delay, avascular necrosis. One case with muscular metabolic myopathy. [89] | Not noted. [89] [91] | Nt noted. [89] [91] | High during flares: ESR, CRP, ferritin, high transferritin, hypercalciuria.[89] Common during flares: metabolic acidosis, aminoaciduria and metabolic abnormalities. Severe sideroblastic anemia, that is extremely microcytic. B cell lymphopenia, variable panhypogammoglobulinemia and/or low levels of mature CD19+ B cells in the blood. Immunodeficiency. [89] Bone marrow biopsy pathology: Erythroid precursors featuring perinuclear mitochondrial iron deposits (“ringed sideroblasts”). [89] | |
| Pediatric Granulomatous Arthritis (PGA) – aka Juvenile Systemic Granulomatosis, Blau syndrome, Early Onset Sarcoidosis, or Jabs Syndrome, NOD2-associated disease-Blau | NOD2-associated Granulomatous Disease | NOD2 (certain mutations) | Autosomal dominant. | Affects all races. | Unknown, but rare. | Intermittent or persistent daily fevers, rash and arthritis. | Rash often develops by 4 months of age, fevers and other symptoms present by 4 years of age. [34] | First symptom: scaly plaques. The rash often starts on the face, then on the torso or chest. Biopsies with non-caseating granulomatous dermatitis. [34] | Intermittent-persistent daily fevers. Some have cranial neuropathies. 80% have vision damage and joint deformities if untreated. Some cases have peripheral nerves affected. [34] | Not noted. [34] | Uveitis (some with blindness). 50% with cataracts, 1:3 patients get secondary glaucoma. Inflamed conjunctiva (conjunctivitis), and/or inflammation of the lacrimal glands, retina and optic nerves is common. [34] | Some have atrial hypertension and/or pericarditis. Some cases with lung involvement. [34] [35] | Hepatomegaly, splenomegaly or hepatosplenomegaly. Some with abdominal pain, higher risk for kidney and/or liver issues. [34] [35] [36] | Splenomegaly, lymphadenopathy. [34] [35] [36] | Symmetrical chronic polyarthritis or oligoarthritis of the wrists, knees, ankles with a boggy appearance is usually caused by an exuberant tenosynovitis. [34] [35] [36] | Some with vasculitis, leukocytoclastic vasculitis. [34] | Not noted. [34] | High CRP and ESR, angiotensin converting enzyme (ACE), immunoglobulins. Anemia, leukopenia, eosinophilia, hematuria, proteinuria, pyuria, abnormal liver function tests (LFTs). [34] [36] | |
| Familial Behçets-like Autoinflammatory Syndrome; Autoinflammatory syndrome, familial, Behcet-like (AISBL); Haploinsufficiency of A20 (HA20) | A20 deficiency; NF-κB regulatory protein A20 (still being classified) | TNFAIP3 | Autosomal dominant.[133] | Unknown. Patients of Turkish, European or Japanese ancestry in the current literature.[133] | Rare. Some families with a few affected members for generations. Some patients were previously diagnosed as having Behçets.[133] | Most cases have symptoms start in childhood with oral and genital ulcers, uveitis and vasculitis that mimic Behçets disease. Some with arthralgia, and/or polyarthritis. A few have periodic fevers. [133] (along with the supplement). | Onset of symptoms in early childhood in most cases, with oral and genital ulcers, uveitis and other findings that present similar to Behçets. One case developed in infancy-most present with their symptoms between 2-16 years of age.[133] (supplement). | Oral and genital ulcers (that look similar to Behçets lesions) axillary and dermal abscesses. [133] Pathergy test is positive in some patients. A few have other skin rashes, such as erythematous papules, folliculitis or a malar rash. [133] (supplement.) | One case with central nervous system (CNS) vasculitis, chorea and migraine headaches. One other patient with headaches. [133] (plus supplement). | Unknown at this time. [133] | Anterior uveitis (may be bilateral), chorioretinal scarring, macular fibrosis (secondary to retinal vasculitis) causing impaired vision. Ocular inflammation is common. [133] (and supplement). | Unknown. One case of pericarditis in infancy. One case of idiopathic thrombocytopenic purpura (ITP) [133] (supplement). | A few with colitis (one case was mild, and undifferenciated). Findings include ulcers in the oropharynx, terminal ileum or colon. One case with nausea, vomiting, weight loss. Inflammation on biopsy of the gastrointestinal tract. [133] (supplement). | A few with lymphopenia, IgG 2, or 2 and 4 subclass deficiencies and low polysaccaride antibodies.[133] (supplement). | Arthralgia, with or without polyarthritis (non-deforming) in the large and small joints. Small joint polyarthritis was the most predominant. Tendonitis in one patient. Some patients have been misdiagnosed as having systemic lupus erythematosis (SLE, Lupus). [133] | Vasculitis, retinal vasculitis. [133] One case with idiopathic thrombocytopenic purpura (ITP). | Unknown. [133] | During flares of symptoms, patients may have elevated CRP, ESR or other inflammatory markers. A few have autoantibodies, or weak positive autoantibodies, such as: Lupus anticoagulant, anti dsDNA, anticardiolipin, RNP, ANA, Some also have HLA-B51, or HLA-B15, or HLA-B39/B44 noted. A few with lymphopenia, IgG 2, or 2 and 4 subclass deficiencies and low polysaccaride antibodies. One case with hemolytic anemia reported, and another with idiopathic thrombocytopenic purpura (ITP). [133] (supplement). | |
| NLRP12-associated Autoinflammatory Disease (NLRP12-AID), NLRP12-Associated Periodic Fever Syndrome – aka Familial Cold Autoinflammatory Syndrome 2 (FCAS2), or Guadeloupe Periodic Fever | NLRP12-associated Autoinflammatory Disease, (Monarch-1) | NLRP12 | Autosomal dominant. Spontaneous mutations, some familial groups. [29] [30] | Unknown. Cases in Guadeloupe, US, Martinique, France, Italy, and Armenia. [38] [39] [75] | Unknown, but rare | 1-3, to up to 7-15 days of fevers 39–40°C, rash and pain. Onset after exposure to cold or cooling temperatures. [38] [39] | Neonatal/early infancy. Rash, fevers, symptoms may be present at birth. [38] [39] | Present during flares: Cold-induced urticaria-like or malar rash noted in some patients. [39] Some with apthous ulcers. [38] [39] Ice cube test is negative. | Fevers 39–40°C myalgia, headaches with flares. Sensorineural hearing loss. Other neurological symptoms are not noted. [31] | Many have increased sensorineural hearing loss. [38] [39] | Not noted. [38] [39] | Not noted. [38] [39] | Some patients have abdominal pain with flares. [39] | Some patients with lymphadenopathy. [39] | Myalgia, arthralgia, fatigue and malaise with flares. Permanent bone or joint damage not noted. [39] | Not noted. [38] [39] | Not noted. [39] | Elevated CRP may be noted during flares. But some patients do not have elevated CRP with flares. [39] | |
| Proteasome-associated Autoinflammatory Syndrome (PRAAS): aka Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature Syndrome (CANDLE), Nakajo-Nishimura Syndrome (NNS), or Japanese Autoinflammmatory Syndrome (JMP) | Proteasome-Associated Autoinflammatory Syndrome (PRAAS) | PSMB8; also PSMB4, PSMB9, PSMA3, POMP [59] | Autosomal recessive. | Unknown. Caucasian, Hispanic, Japanese patients, and one case in South Africa. [27] [60] | Unknown, but rare. | Chronic-frequent fevers with disease flares. Inflammatory flares and symptoms are often present before the age of 6 months. [26] [27] | Onset at birth or in infancy. Progressive damage from chronic inflammation noted as the child grows. [26] [27] | Annular cutaneous plaques with residual purpura. Lipodsystrophy first noted on the face and around joints, and the limbs. Lips swell with flares. Persistently swollen, violaceous (purple-red) eyelids. [26] [27] | Aseptic meningitis and systemic inflammation. Growth delays are common, with low height and weight. Developmental delays noted. [26] [27] | Some have frequent otitis and/or recurrent sinusitis. [27] | Nodular episcleritis (inflammation on the eye). Conjunctivitis. Keratitis. Periorbital edema. Chronically swollen, violaceous (purple-red) eyelids. [26], [27] | Clubbing of the fingers and/or toes. At risk for cardiac arrythmias and dilated cardiomyopathy. [26] [27] | Diarrhea or loose bowels with flares. Hepatomegaly and splenomegaly (hepatosplenomegaly) with elevated liver enzymes. Delayed or slow growth. At risk for being underweight, or failure to thrive. At risk for multi-organ failure and death. [26], [27] Large abdomen from an enlarged liver and spleen is possible. | Splenomegaly, lymphadeopathy. [26], [27] | Joint contractures, muscle atrophy, panniculitis induced lipodystrophy, myositis, fatigue and malaise. Inflamed nose and ear cartilage (chondritis). Short stature. Delayed growth in height and weight is common. [26] [27] | Not noted. [26] [27] | Not noted. [26] [27] | Hypochromic or normocytic anemia. High CRP, ESR, triglycerides. Some with elevated platelets, TSH, and/or LDL. [26] [27] | |
| Spondyloenchondrodysplasia with immune dysregulation (SPENCDI); aka Roifman immunoskeletal syndrome combined immunodeficiency with autoimmunity and spondylometaphyseal dysplasia; or Roifman-Costa Syndrome | Interferon Mediated Autoinflammatory Diseases | ACP5 | Autosomal recessive. | Not noted. | Unknown-rare | Recurrent respiratory infections, bone and cartilage deformities, low-set ears, pointy nose noted early in life. [108] [109] | Onset of symptoms in infancy, childhood or adolescence. [108] [109] | Hyperpigmented macules, vitiligo. Narrow, pointy nose. Low-set ears. High risk for severe varicella infection. [109] Some with symptoms of Raynaud's. [111] One case with Sjögren syndrome-like symptoms and severe scleroderma. [112] | Most patients do not have neurologic involvement. One case of fatal encephalitis. [109] Some cases with spasticity, cognitive delay or deficits, and/or developmental delay, and cerebral calcifications (late-onset). [109] [110] | Normal hearing. Recurrent or frequent otitis media. Low-set ears. [109] | Not noted [109] [110] | Recurrent respiratory infections (sinusitis, pneumonia), restrictive lung disease, and/or interstitial fibrosis. Idiopathic thrombocytopenic purpura (ITP). [109] | Some have Crohn's disease, and diarrhea. One case of entercolitis. Patients are at risk for concurrent autoimmune diseases. Hypothyroiditis. [109] | Lymphadenopathy. [109] Idiopathic thrombocytopenic purpura (ITP), symptoms of systemic lupus erythematosis (SLE) and other autoimmune diseases. [109] | Short stature. Bone deformities: Spondylometaphyseal dysplasia. Irregular, sclerosed bilateral distal metaphyses of the radius/ulna, femur, and proximal fibula. Spinal defects: platyspondyly, irregularities in the vertebral endplates. [109] Cartilage is affected (ears and nose too). [108] Some with concurrent Juvenile Rheumatoid Arthritis (JRA). One case with polymyositis. [112] | Not noted. [109] [112] | Not noted. [109] [112] | Normal or elevated IgG levels. Decreased circulating T cells. Hypothyroidism. Idiopathic thrombocytopenic purpura (ITP). Humoral and celular immunodeficiency. [109] | |
| STING-associated vasculopathy with onset in infancy (SAVI);TMEM173-AID | Interferon Mediated Autoinflammatory Diseases | TMEM173 | Autosomal dominant. | Unknown | Unknown-rare | Continuous, starting in infancy with fevers and progressive skin lesions and vasculitic changes. Cold-induced flares. [98] | Infancy-before 8 weeks in most cases. [98] | Cold-induced flares. [98] Relapsing malar rash, thin hair. [93] Scaling, pustular, violaceous and/or blistering rashes on the distal fingers, toes, nose, cheeks, & pinnae of the ears that develops into acral necrosis and/or gangrene (leading to amputation of digits). Chilblains. Eschar and painful crusts on ulcerated lesions. Skin lesions worsen in the winter. [92] Skin biopsy: Inflammatory infiltrate of the dermis plus signs of leukocytoclastic vasculitis & microthrombotic angiopathy of the dermal small vessels and leukocytoclasia. Also deposits of IgM or vessels with fibrin. [82] One case of fatal necrotizing facscitis. Telangiectasias, Raynaud's, livedo reticularis, perforated nasal septum. [98] Nail loss or dystrophy. [93] | Rare: basal ganglia calcifications. Normal cognitive function.[98] | Not noted [98] | Not noted [98] | Interstitial lung disease and pulmonary fibrosis noted on CT scan in many patients, that in some cases can be fatal. [82] [93] Emphysema [98] Tachypnea at birth [92], abnormal pulmonary-function test (PFT). A few with arterial hypertension. [98] Hilar or paratracheal lymphadenopathy, mixed, scattered lymphocytic inflammatory infiltrate on a lung biopsy. [82] | Failure to thrive is common. [82] Abdominal, liver or spleen issues are uncommon. [98] | Occasional general lymphadenopathy. [98] Hilar or paratracheal lymphadenopathy noted [82] | Seropositive polyarthritis. [82] Arthralgia, myositis. [98] | Widespread vasculitis. [82] Damaged small vessels are common, plus nailfold capillary tortuosity and the loss of capillary loops. Telangiectasias noted on the limbs and hard palate. [82] | Not noted [82] | High: ESR, CRP, IgG, IgA. Hypergammoglobulinema. Anemia, leukopenia, thrombocytosis, T-cell lymphopenia with normal B cells. [92] Normal or low positive ANA, c-ANCA, p-ANCA, antiphospholipid antibodies that later disappear, or are variable. [82] [92] [93] | |
| Aicardi-Goutieres syndrome(s) (subclassifications 1-7); aka Familial infantile encephalopathy, with calcification of basal ganglia and chronic cerebrospinal fluid lymphocytosis | Interferon Mediated Autoinflammatory Diseases | Neonatal onset, high mortality: TREX1 (AGS1), RNASEH2A (AGS4), RNASEH2C (AGS3). Later onset, lower mortality: IFIH1 (AGS7), DSRAD (AGS6), SAMHD1 (AGS5), RNASEH2B (AGS2) | Autosomal recessive in most cases. TREX1 and DSRAD can also present as autosomal dominant. | Not noted. | Unknown-rare | Episodes can last for months with the onset of symptoms and encephalopathy, in the first year of life. 40% have chilblains exacerbated by the cold, starting after 1 year of age. [106]. Some with continuous symptoms from birth. [104] | Encephalopathy is often the first symptom noted in the first year of life, with fevers, irritability and progressive neurologic changes with flares of symptoms. 40% of patients develop chilblains lesions, starting in early childhood. 20% of cases have AGS symptoms at birth, but most are asymptomatic at birth. [104] 25% Die within the first year of age. [104], [105] | Chiblains are the classic skin symptom seen in 40% of patients with AGS, and may be start to develop after the first year of life. [106] They can be cold-induced and/or exacerbated by cold exposure. Chilblains can be painful, pruitic, swollen, and present on the distal areas of the ears, fingers and toes. There is a range of severity, from cold fingers and toes, and/or erythematous plaques to severe tissue damage and auto-amputation. [106] Microvascular vasculitis noted. [104], [106]. May present with purpura, petechiae, acrocyanosis. Neonatal jaundice in some patients. [105] Some have aphthous ulcers [104] | Fever lasting days or weeks during attacks of symptoms, with Irritabiliity. Some have seizures. Progressive encephalopathy and cerebral atrophy, mental delay and damage, starting in infancy. [104], [105], [106] Worsening microcephaly. [105]. Worsening cognitive and mental regression with deficits, that can progress to a persistent vegetative state, or death. Dystonia, Low IQ [105]. Calcifications in the white matter, or basal ganglia noted. [105]. Leukoencephalopathy. Lymphocytosis in the CSF. [105] | Not noted. [104], [105], [106] | Progressive eye involvement due to neurologic damage. [105]. Glaucoma, and/or cataracts in some cases. [106] Visual inattention. [104], [105], [106] and ocular jerking in some cases. [107] | Not noted. [104], [105], [106] | Vomiting, feeding difficulties are common. [107] Uncommon: hepatomegaly, splenomegaly (hepatosplenomegaly) [104], [105], [106]. | Some cases with splenomegaly. [105], [106] | Some with stiff joints. Spasticity, dystonia of the limbs or hypotonia of the trunk noted. [104] | Vasculitis in the small vessels noted on skin biopsy of distal lesions from chilblains. [106] | Not noted. [104], [105], [106] | High: Liver enzymes, abnormal liver function tests (LFTs). CSF lymphocytosis, with increased serum and CSF alpha interferon levels. [104]. Low: Thrombocytopenia, Normal: ESR, CRP, ANA, lupus anticoagulant, Anticardiolipin, antiphospolipid, IgG, IgM, cryoglobulins, alpha-1 antitripsin. [106] | |
| Sweet's Syndrome (SS); aka Acute Febrile Neutrophilic Dermatosis | Neutrophilic Dermatoses | Unknown at this time. More common in carriers of HLA-B54. | Unknown | Affects all races. | Rare. One study found the incidence to be 3 per 10,000 amongst new dermatologic patients in Iran. [99] | Fevers, often for many days-weeks. Fever may preclude or happen at the onset of other SS symptoms. [97] 1/3 to 2/3 of patients have recurrent Sweets symptoms. Other cases involve a single episode of symptoms in their lifetime. Three forms: Classical/idiopathic SS, drug-induced (DISS), or malignancy-associated Sweet's syndrome (MASS). [97] | Adulthood, more frequently in middle-aged women, but also in men. It rarely presents in infants or children. [96] Sweet's presents in response to some trigger, such as infection, inflammation (IBD/Crohns/UC), autoimmune diseases, vaccinations, cancers, or certain medications. [96] | Cutaneous pathergy. Multiple painful and tender skin lesions varying from papules and vesicles to wide, thick, swollen plaques. The most common areas for the rash is on the neck, face, upper chest, back and limbs. Some have sores only in areas that are exposed to the sun. [96] Lesions can be annular (ringed), pseudovesicular, and may look like atypical pyoderma gangrenosum. [96] Apthous, or mucosal ulcers (even ocular ulcers at times). [96] Some have erythema nodosum. [97] Skin biopsy: Neutrophillic inflammation and leukocytoclasia without vasculitis. MASS cases may also present with additional findings like leukemia cutis. [96] [97] Subcutaneous Sweet's affects deeper tissues and may resemble cellulitis. [96] Neutrophillic dermatosis of the hands (variant Sweet's) causes violaceous nodules, mostly on the top of the hands and digits than on the palms. [96] Histocytoid Sweet's is associated with malignancies or rheumatoid arthritis, with myeloid cell infiltration on the biopsy instead of neutrophils. Nodules, lesions, or plaques are oval, red to violet or brownish. [96] | Fevers lasting many days to weeks before the onset of symptoms and during flare. [97] (Fever may be absent in MASS presentations). Headache, fatigue and malaise are common. [96] Neuro-Sweet's disease (rare). A few cases with CNS involvement, aseptic meningitis, and/or encephalitis. [97] | Some cases with pustules or plaques in the exterior auditory canal and the tympanic membrane. A few cases with sensorineural hearing loss (in Neuro-Sweet's). [97] | Ocular Sweet's Syndrome can affect vision, presents with painful, "sticky" eyes that can develop ocular ulcers. [96] At risk for conjunctivitis, conjunctival hemmorhage, blepharitis, iritis, uveitis, keratitis, episleritis, scleritis, acryoadenitis, choroiditis, periocular swelling, and/or glaucoma. [96] [97] | Respiratory infection is a common precursor to the onset of idiopathic SS symptoms. [97] Blood cancers, most commonly acute myelogenous leukemia (AML) can lead to an onset of MASS. [97] Some cases with concurrent aortic stenosis, cardiomegaly, vascular dilation, neutrophillic myocardial infiltration, and heart failure, or other cardiac conditions. [97] A few cases with pharangeal mucosal infiltration, swelling and upper airway obstruction, pleural effusion, bronchial pustules and inflammation. [97] | Higher risk for inflammatory bowl diseases (IBD) such as Crohns and ulcerative colitis. [96] 11-50% of idiopathic SS have abnormal renal function. [97] Pregnancy can trigger idiopathic SS [96] [97] Hepatomegaly, splenomegaly in some cases. Some cases with: neutrophillic inflammation in the intestines, or in the hepatic portal. Inflammatory bowel disease (IBD), Crohns, or ulcerative colitis may trigger Sweet's. Hematuria, proteinuria, glomerulonephritis. [97] | Splenomegaly. Some with lymphadenopathy. [97] | Arthralgia, arthritis. Some with relapsing polychondritis. Sweet's may present in patients that also have Rheumatoid Arthritis (RA), Systemic Lupus Erythematosis (SLE), Stills (AOSD) or sarcoidosis. [96] Some cases with osteitis, or CRMO/SAPHO. Children may have dermatosis-related sterile osteomyelitis. [97] | Absent in Sweet's. [96] | Not noted. [96] [97] | High (when symptomatic): ESR, CRP, neutrophil leukocytosis, p-ANCA or c-ANCA is sometimes noted. If associated with a blood cancer or other malignancy there are altered (high or low) levels of RBCs, WBCs, platelets, or bone marrow biopsy findings. [96] | |
| Behçets Disease | Idiopathic, Neutrophilic Dermatoses | ERAP1 (with HLA-B51); also variants near: CCR1, KLRC4, STAT4. [42] Genetic testing in research studies only. (Article with more info in the Genetic testing button) | Complex | Rare in the USA. More common in the Middle East, Asia and Japan (Silk Road Route). [42] [43] | Prevalence is 80-370:100,000 people in Turkey, 10:100,000 in Japan & 0.6:100,000 in Yorkshire, UK. [3] | Aphthous ulcers are present in almost all patients during flares, for around 10 days with inflammation in the eye and arthritis. [42] [43] | Most show symptoms in early adulthood (20s-30s) but the onset can be in childhood, or any age. [42] [43] | Pathergy. Pseudofolliculitis, erythema nodosum-like and/or acneiform nodules. 98% with aphthous ulcers & 65% have genital ulcers. [43] | 20-40% have Neuro-Behçets with headaches, aseptic meningitis or meningoencephalitis, seizures, hemiplegia, or cranial nerve palsies. Cerebral venous thrombosis with high intracranial pressure (ICP) noted. [43] | Not noted. [42] [43] | Frequent anterior and/or posterior uveitis. Cataracts, retinal vasculitis <30% risk for blindness. Papilledema with neurologic involvement (Neuro-Behçets). [43] | Myocarditis, endocarditis with aortic or mitral insufficiency, arterial aneurysm, pulmonary embolism. [43] | Ulcers from mouth to anus. Some with hepatomegaly, splenomegaly or hepatosplenomegaly. Nausea, abdominal pain, anorexia, diarrhea (may be bloody). [43] | Splenomegaly; lymphadenopathy. [43] | 45% have arthralgias and/or arthritis–often the knees and/or ankles, but other joints can be affected. Joint issues may be the first sign of Behçets. X-ray is normal but the synovium often has high neutrophils or mononuclear cells and a vasculitis process. [43] | Extensive vasculitis. 30% with venous thrombosis. [43] | Not noted. [42] [43] | Leukocytosis common. Normal–rarely elevated ESR or CRP. Some cryoglobulinemia, elevated factor VIII, fibrinolysis. [43] | |
| Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Cervical Adenitis (PFAPA) – aka Marshall Syndrome | Idiopathic | Currently unknown. No genetic testing available. | Currently unknown. | Affects all races. [40] | Unknown. PFAPA is the most common non-infectious recurrent fever disorder. [40] | Periodic fevers and symptoms lasting 3-6 days, recurring every 21-28 days. Extremely predictable frequency of days between the onset flares in most cases. Patients are symptom-free between flares. No specific triggers have been identified–flares come at regular intervals, often monthly. [40] If there are any persistent symptoms between flares, evaluation for autoinflammatory periodic fever syndromes is essential. | Early childhood, usually between 2-5 years of age. A few adult-onset cases are documented. Most teens outgrow it. [40] | Aphthous ulcers (stomatitis), and pharyngitis with exudate, (but no infection) is a classic finding. [40] [41] Skin rashes are not typical, or a part of the PFAPA diagnostic criteria-if rashes occur with flares, or are persistent between flares, other autoinflammatory diseases should be considered. | High fevers for 3-6 days, with chills and malaise. Some patients have headaches with flares. Other neurological symptoms are not noted. [41] | Not noted. [40] [41] | Not noted. [40] [41] | Flares of fevers, stomatitis and pharyngitis are not associated with respiratory illness. [40] [41] | Abdominal pain, and diarrhea are often present with flares. [40] [41] | Cervical adenopathy or lymphadenopathy during flares. [40] [41] | Arthralgias, fatigue and malaise. No permanent joint or bone issues noted, and patients are symptom-free between PFAPA flares. [40] [41] | Not noted. [40] [41] | Not noted. [40] [41] | High: ESR, CRP, WBC during flares only. All normal labs when not flaring, or between flares. [40] [41] IgD may be elevated in patients with PFAPA (and these patients were negative for MVK mutations). [126] PFAPA symptoms overlaps with a number of other autoinflammatory periodic fever syndromes, so genetic testing and attention to clinical findings may be helpful. | |
| Systemic-Onset Juvenile Idiopathic Arthritis – aka Still’s, Systemic Juvenile Idiopathic Arthritis | Macrophage Activation Diseases | Currently unknown for most cases. LACC1 noted in some patients. [100] HLA-DRB1 in some patients with European ancestry. [57] Genetic testing in research only at this time. | Complex | Affects all races. soJIA accounts for 10% of all JIA. [45] | Uncommon. 0.4–0.9 cases per 100,000 people, per year. [46] | Fevers often > 39°C 1-2 times/day for >2 weeks, most often occurring in the evening with arthralgia, rash and other symptoms. [45] [46] | Onset before the age of 16–most often by 2 years of age, or between 0-5 yrs. of age. [46] | Rash: Fleeting, evanescent, migratory, bright salmon-pink, morbilliform, macular rash that often presents with the onset of fevers. [45] [46] | High fevers >39°C 1-2 times/day for >2 weeks. Other neurological symptoms are rare. A few cases with seizures, meningismus, irritability and decreased level of consciousness. [46] | Not noted to be from soJIA. [45] [46] | Uveitis can be a complication from soJIA. [46] | Serositis (especially pericarditis) is often seen. Pleuritis (pleurisy), pleural effusions can occur. Some with pulmonary disease. Risk for Macrophage Activation Syndrome (MAS). [46] | Peritonitis rarely occurs. 50% have an splenomegaly, some with hepatomegaly. [46] | Many with generalized lymphadenopathy. Some with mesenteric adenitis. Half with splenomegaly. [45] [46] | Arthralgias may come before the arthritis. 88% have polyarticular or oligoarticular arthritis, most often in the wrists, knees, and/or ankles. Some with cervical spine, hip, temporomandibular joint arthritis or synovial cysts. [45] [46] | Not noted. [45] [46] | Amyloidosis occurs in 7.4% of patients in the USA, and 16% in Turkey. [46] | High: ESR, CRP, WBC, SAA, ferritin, aldolase, IL-18. Elevated liver function tests (LFT’s). Leukocytosis, thrombocytosis. Anemia is common. [45] [46] | |
| Adult-Onset Stills Disease – aka Adult Still’s, Wissler-Fanconi Syndrome | Macrophage Activation Diseases | Currently unknown. No genetic tests are available at this time. | Currently unknown. | Rare. Affects all races. [44] | France: estimated that 0.16:100,000 people have AOSD. AOSD affects more women than men. [44] | High fevers > 39°C that last for <4 hours, recurring more than once a week, with a maculopapular rash and arthralgia. [44] | First onset of symptoms occurs between 16-35 years of age. Affects all ages. [44] | Evanescent, salmon-pink, mildly pruritic maculopapular rash on the proximal limbs and trunk. [44] | >95% have high, spiking fevers, fatigue and myalgia with flares. Other neurological symptoms are very rarely seen. [44] | Not noted. [44] | Not noted. [44] | <25% have pleuritis (pleurisy), pericarditis (a few with tamponade). Some myocarditis, pleural effusions, Adult Respiratory Distress Syndrome (ARDS). [44] | 50-75% with hepatomegaly, abnormal liver function tests (LFTs). 43% with splenomegaly. Some with hepatosplenomegaly. Renal disease is rare. [44] | Lymphadenopathy is common. Many with splenomegaly. [44] | Myalgias, arthralgias and/or arthritis are common. Wrist changes after 6 months. 41% develop intercarpal and carpometacarpal joint space narrowing a few years after onset of AOSD – 25% then develop pericapitate ankylosis. [44] | Not noted. [44] | Very rare. [44] | High: ESR, CRP, LFTs, Ferritin, IL-18. Low glycosylated ferritin. Leukocytosis, anemia common with flares. Prolonged PTT: risk for disseminated intravascular coagulation (DIC). [44] | |
| NLRC4-associated Autoinflammatory Disease, NLRC4-associated Macrophage Activation-like Syndrome (NLRC4-MAS) | NLRC4-associated autoinflammatory diseases, Macrophage Activation Diseases | NLRC4 (aka SCAN4, CARD12) | Autosomal dominant. | Not noted. | Unknown-rare | Injury, surgery or other stressors (physical or emotional) can trigger systemic flares of fever and macrophage activation syndrome (MAS)-like symptoms that can persist for weeks. [95] | Infancy, early childhood - some at birth. [94] [95] | Evanescent rash with occasional dermatographism, with a notable urticarial rash during disease flares. [94] Adult with seronegative psoriatic arthritis, and erythematous plaques. [95] | Recurrent fevers, starting in early infancy. [94] [95] Uncommon for other CNS symptoms. [98]. | Not noted [94] [95] [98] | Not noted [94] [95] [98] | Risk for multiple-organ failure, disseminated intravascular coagulation (DIC), macrophage activation syndrome (MAS)-like attacks, with Acute Respiratory Distress Syndrome (ARDS). [95] | Failure to thrive, or challenges with weight gain is common. Vomiting and diarrhea during flares. Splenomegaly, transaminitis. Inflammatory infiltrates are noted in the intestine. Duodenitis, neonatal-onset enterocolitis noted (may resolve after 1 yr of age). [94] [95] [98] High risk for multiple organ failure with MAS-like attacks. [95] | Splenomegaly.[94] [95] Lymphadenopathy may occur. [98] | Short stature, low weight in one case. [95] Arthralgia, myalgia, and psoriatic arthriitis noted in an adult patient. [95] | Not noted [94], [95] | Not noted [94], [95] | High (during flares): CRP, ESR, IL-18, triglycerides, ferritin (extreme ferritinemia), ALT, AST. Over-production of IL-1β and IL-18. Low during MAS-like flares): hemoglobin, platelets (thromobocytopenia), anemia, leukopenia. Normal NK cell function. [94]. | |
| (Primary) Familial Hemophagocytic Lymphohistiocytosis– aka Familial Erythrophagocytic Lymphohistiocytosis | Macrophage Activation Diseases | PRF1, STX11, STXBP2, MUNC13-4 , RAB27A, UNC13D; X linked: SH2D1A, | Autosomal recessive, but if X-linked: inheritance is dominant. | Affects all races. 80% of African Americans and 20% of patients with European decent have PRF1 mutations. [47] [48] | 1° HLH affects 1:50,000 people worldwide. [48] | Fevers often > 39°C 1-2 times/day for >2 weeks, most often occurring in the evening with arthralgia, rash and other symptoms. | Onset <1yr: often by 6 months to early childhood. Some in utero or late childhood cases. A few adult-onset cases. [47] [48] [49] | 40% with transient maculopapular, nodular or purpuric skin rashes during bouts of high fever. Jaundice. [47] [48] | High fevers. Increased CSF protein. High intracranial pressure (ICP). Multifocal inflammation of the gray and white matter, intracranial bleeding, generalized atrophy or brain edema, seizures and/or coma. [47] | Not noted. [47] [48] [49] | Blindness due to brain inflammation. [48] | High risk for respiratory infections triggering fevers, edema, systemic inflammation and macrophage activation syndrome (MAS). [49] | Hepatomegaly, splenomegaly, or hepatosplenomegaly. Liver disease (hepatitis) is common. High risk of death from multi-organ failure in 2+ months if untreated. [49] | Lymphoma. Hemophagocytosis– spleen and lymph nodes. Splenomegaly. [49] | Hemophagocytosis in the bone marrow. Delayed closure of the bones of the skull in infants, with a bulging fontanel often noted. Neck stiffness, abnormal muscle tone, impaired muscle coordination, paralysis. [48] [49] | Not noted. [47] [48] [49] | Not noted. [47] [48] [49] | High: ESR, CRP, triglycerides, liver Function tests (LFTs), especially ALT, soluble CD25, ferritin, sIL2r. Low: platelets, fibrinogen. Low NK cell cytotoxic function, neutropenia, anemia. [49] | |
| X-linked familial hemophagocytic lymphohistiocytosis; XIAP deficiency; X-linked lymphoproliferative syndrome type 2 (XLP-2)-MAS | Macrophage Activation Diseases | XIAP (BIRC4) | X-linked. X-linked dominant male inheritance (typical presentation). There are some female carriers with some symptoms. Testing recommended for XIAP (and also SAP) in males with symptoms of HLH after Epstein-Barr virus (EBV) infections, or HLH presenting in childhood or adolescence. [84] | Unknown | Unknown-rare | Onset of HLH may be the first sign of this disease, and may occur after Epstein-Barr infections (that can be severe). [85] Some cases developed pancytopenia and splenomegaly after MMR vaccine. [88] | Infancy to early childhood. A few with school-age onset. [84] [88] | Some patients may have erythema nodosum, including female carriers of an XIAP deficiency. [85] One case of recurrent rashes with fevers. [83] | Prolonged, periodic fevers >7 days. [84] A few with pathologic cerebrospinal fluid (CSF). [85] | Uncommon. One case with sensorineural hearing loss in childhood. [83] Hearing loss not noted in other cases. [84] [85] | Uveitis [83] [87]. One case with bilateral cataracts in early childhood. [83] | Severe cases of infectious mononulcleosis leading to HLH is common. [84] [85] Some with increased risk for severe, recurrent or rare respiratory infections. [88] | Higher risk for early-onset inflammatory bowel diseases (Crohns, colitis, IBD), even in female carriers of the XIAP deficiency mutation. [85] Hepatomegaly, splenomegaly, granulomatous hepatitis. [83] [88] Chronic and/or hemorrhagic colitis that can be fatal (one case with portal hypertension). [88] Crohns-like disease, celiac-like disease. [87] Some patients with chronic liver failure and/or hepatitis. [85] Cholangitis (sometimes associated with colitis). [88] | Splenomegaly (may be the first presenting symptom). [88] Hemophagocytic lymphohistiocytosis (HLH) affects 90% of cases [85] and many can have recurrent HLH. HLH can occur after Epstein-Barr virus (EBV). [85] [88] No known patients with lymphoma or aplastic anemia. [84] | Uncommon. [84] [85] [88] One case with arthritis in the hips and knees that also developed myositis. [83] | Not noted. [83] [84] [88] | One case with elevated serum amyloid. [83] Not tested in other cases in the literature. | High: ESR, CRP, liver function tests (LFTs), triglycerides, ferritin, [85] IL-18 [86] cytopenia. [85] Low: fibrinogen, platelets, anemia, neutropenia, hemophagocytosis. [88] Normal NK cell function [85] Variable NKT cell defects. [84] IgG normal in untreated patients. [85] Low IgG secondary to treatments. [84] [85] | |
| PLCG2-associated Antibody Deficiency & Immune Dysregulation (PLAID) – aka Familial Atypical Cold Urticaria (FACU) or FCAS3 | PLCG2-associated | Heterozygous genomic deletions within the PLCG2 gene. [64] | Autosomal dominant. | Unknown. Most reported cases with European ancestry. | Unknown but rare. | Onset <5 minutes after exposure to cold air (evaporative cooling). [64] Frequent sinusitis or pneumonia, and respiratory infections. Concurrent autoimmune diseases. [64] [65] | Onset in infancy-under 6 months of age. Lifelong symptoms, but some find the symptoms less severe in adulthood. [64] [65] | Cold-triggered. Cold urticaria, erythema and itching post cold exposure (air, wet skin, cold food). Some w/angioedema; chronic granulomata. Some with ulcerative and cutaneous lesions from cold exposure.[132] Negative Ice cube test. [64] | Not noted. [64] [65] No fevers noted with cold-induced urticaria. [64] [65] | Not noted. [64] [65] | Uncommon. [66] | 44% with recurrent sinus and/or respiratory infections, >50% with allergies, asthma and/or autoimmune diseases. [65] | Not noted. Some have concurrent autoimmune diseases that may involve other organs. [65] | Not noted. Some need IVIG for low immunoglobulins & frequent infections. A few with combined variable immune deficiency (CVID). [65] | Not noted. [64] [65] Some have concurrent autoimmune diseases that may involve the joints, such as inflammatory arthritis or undifferenciated connective tissue diseases. [65] | Not noted. [64] [65] | Not noted. [64] [65] | High IgE. Low serum IgA, IgG, IgM. Decreased circulating CD19+ B cells, IgG+ & IgA+ memory B cells, NK cells. >60% +ANA. WBC normal. [64] [65] | |
| Autoinflammation & PLCG2-associated Antibody Deficiency and Immune Dysregulation (APLAID) | PLCG2-associated | PLCG2 mutation | Autosomal dominant. | Unknown | Unknown but rare. | Recurrent skin lesions, chronic inflammation, and progressive eye complications from this disease. [66] [67] [68] | Onset in infancy with recurrent skin lesions, arthralgia, eye inflammation, and infections. Some with intestinal symptoms. [68] | Erythematous plaques and vesiculopustular blistering rash that intensifies with heat & sun exposure. Cellulitis often develops with rashes. [66] [67] | Not noted. [66] [67] [68] No fevers noted as a part of this disease (but fevers may be present with infections). [66] [67] [68] | Not noted. [66] | Many develop corneal erosions, blisters, ulcerations, intraocular hypertension, and/or cataracts. [66] [67] [68] | Mild humoral immunodeficiency with increased frequency of sinus and/or respiratory infections or interstitial pneumonia. [66] [67] | Some with bouts of abdominal pain, bloody diarrhea, enterocolitis, or ulcerative colitis. [67] [68] | Not noted. [67] [68] | Not noted. [67] [68] | Not noted. [67] [68] | Not noted. [67] [68] | Low circulating IgA, IgM antibodies, decreased class-switched memory B cells & NK T cells. ANA negative. [67] [68] | |
| Lyn kinase-associated Autoinflammatory Disease (LAID) | Increased Immune Cell Receptor Signaling | LYN | Autosomal dominant. | Not noted. | Unknown-rare. | Symptoms present at birth, and continuous. [102] | Symptoms may be present at birth or early infancy. [102] | Purpuric rash presenting at birth or in infancy. Vasculitis noted on skin biopsy. Periorbital erythema, and testicular swelling in one case. [102] | Unknown [102] | Unknown [102] | Periorbital erythema. [102] | It is unknown, or not noted if there are any symptoms affecting the heart or lungs. Hydrops fetalis in one case. [102] | Hepatomegaly, splenomegaly (hepatosplenomegaly). One case with testicular swelling. One case needing a splenectomy for refractory thrombocytopenia and the patient developed periportal lymphocytic infiltrates with a vanishing bile duct disorder. Hydrops fetalis in the same patient at birth. [102] | Splenomegaly. Splenectomy done for refractory thrombocytopenia in one case. [102] | Unknown [102] | Skin biopsy: vasculitis noted. [102] | Not noted. [102] | High: CRP, elevated liver enzymes, leukocytosis, anemia, thrombocytopenia (severe). Positive for autoantibodies, such as +ANA, anti-Sm, anti-SSA, antiphospholipids and anti-mitochondrial antibodies. [102] | |
| SH3BP2 deficiency with multiocular cystic disease of the mandibles (SDCM), aka Cherubism, | Increased Immune Cell Receptor Signaling | SH3BP2 | Autosomal dominant. | Not noted. | Unknown-rare. Affects more males than females. [114] | Continuous and increasing jaw growth (usually painless) during early childhood. [114] | Onset of persistent and increasing jawbone growth between 14 months to 3 years of age. Most have slower growth of the jaw by 5 years of age, and the process halts by 12-15 years of age, and the bone changes resolve in adulthood. [114] | No rashes or symptoms affecting the skin noted. Cherubism may be seen in some other conditions, such as neurofibromatosis that are associated with some skin findings. [114] | No characteristic symptoms from Cherubism itself, but cherubism may also present in other syndromes, such as Ramon or Noonan syndrome that may have CNS symptoms. [114] | Rare. A few extreme cases of deformed jaw growth affecting the ear region, which can impact hearing. [114] | "Eyes to heaven" turned up eyes showing the sclera below the iris in some patients. A few cases with orbital bone involvement which can affect vision. [114] | A few cases with obstructive sleep apnea, or gingival fibromatosis. [114] Extreme cases of jaw enlargement may cause dental issues . [114] If presenting with Noonan synrome, cardiac defects may be noted. [113] | Not noted to be caused by cherubism. [113] [114] | Cervical lymphadenopathy. [113] [114] | Onset in early childhood-increased jaw size leading to a "puffy or chubby cheek" appearance due to painless mandible and maxilla enlargement from cysts with fibrous dysplasia. Jaw growth stabilizes during puberty, and facial features improve and are normal in most adults with the disease. [113] Can affect the development of the teeth. Alveolar cavity damage can displace the teeth, and dental x-rays may appear as "floating teeth syndrome". [114] Rarely affects long bones. [115] X-ray: bilateral multilocular radioleucent areas. Histopathology: fibrous connective tissue with large numbers of multinucleated giant cells in the background. [114] | Not noted. [113] [114] | Not noted. [113] [114] | CRP, ESR may be normal or slightly elevated. REF#115 | |
| SLC29A3 Spectrum Disorder – aka H. syndrome; Pigmented Hypertrichosis with Insulin-dependent Diabetes Mellitus (IDDM); Faisalabad Histiocytosis and Sinus Histiocytosis with Massive Lymphadenopathy | SLC29A3 related | SLC29A3 | Autosomal recessive. | Unknown. Many patients with Middle Eastern ancestry. Some from India, Pakistan, Spain, Bulgaria. [70] | Unknown but rare. | Fever 39°C with flares that last 7-10 days with joint and abdominal pain; pericarditis and sometimes diarrhea. Flares occur once every 2-3 months. [70] | Onset in infancy-starting with recurrent fevers and flares. Chronic and progressive systemic symptoms develop. [69] [70] [71] | Hyperpigmentation with hypertrichosis. [69] [70] [71] [72] Some with notable varicose veins on the legs. [69] | Fever 39°C with flares lasting over a week. [69] [70] Psychomotor impairment or delays. Dysmorphic facial features noted. [69] [70] | Sensorineural hearing loss, from early infancy or childhood. [69] [70] | Uveitis. Vision loss or blindness can occur from anterior uveitis and glaucoma. Ptosis, eyelid swelling from histiocytic deposits are noted. [69] [70] [71] | Pericarditis with flares. Cardiac defects noted: ASD, VSD, PDA, mitral valve prolapse, cardiomegaly and other findings. [69] [70] | Diabetes Mellitus. Hepatosplenomegaly. Abdominal pain, diarrhea, failure to thrive. Hypogonadism. [69] [70] | Lymphadenopathy. Rosai-Dorfman sinus histiocytosis with massive lymphadenopathy. [69] [70] [71] | Short stature. Arthralgias. Dysmorphic facial features: triangular face, rotated ears, macrocrania, exophtalmia. Pectus excavatum, wide-set nipples, widened ribs, long bone changes, short, square hands, sacrococcygal dimple, contractures. [69] [70] | Not noted. Some with varicose veins on the legs. [69] | Not noted. [69] [70] [71] [72] | Chronically elevated, but increase during flares: CRP, ESR, WBC. High during flares: IgG, Ig A. Anemia. [69] [70] [71] [72] | |
| Deficiency of Adenosine Deaminase 2 (DADA2) – aka Fever with Early Onset Stroke (FEOS) | ADA2 deficiency | ADA2 (aka CECR1) | Autosomal recessive. | Unknown. | Unknown but rare. | Intermittent, recurrent fevers, livedo reticularis rash, vasculopathy, and a high risk for early-onset lacunar stroke. [73] [74] | Onset of symptoms in infancy–early childhood with recurrent fevers, livedo reticularis rash, and vasculopathy. [73] [74] | Livedo reticularis rash is common. A few with polyarteritis nodosa (PAN). Diffuse vasculopathy, with impaired endothelial integrity and endothelial cellular activation. [73] [74] Predominant levels of neutrophils and macrophages in the interstitium; perivascular T lymphocytes, without obvious vasculitis. [131] | Recurrent fevers, and early-onset lacunar strokes is common. Some patients do not have strokes. [131] Possible adult stroke risk. Brain biopsies: diffuse vasculopathy, with impaired endothelial integrity and endothelial cellular activation. [73] [74] The first stroke often occurs before 5 years of age. [131] | Unknown. [73] [74] | Unknown.[73] [74] Strokes have the potential to cause blindness. | Unknown. [73] [74] | Hepatosplenomegaly; diffuse vasculopathy noted in the liver. [73] [74] Portal hypertension in some patients. [131] | Not noted. [73] [74] | Not noted. [73] [74] | Diffuse vasculopathy in the skin, liver and brain. [73] [74] No signs of cerebral vasculitis noted. One case of necrotizing vasculitis [131] | Not noted. [73] [74] | High: CRP, ESR with flares. Cytopenia. Blood: 10-fold decrease in ADA2. Low: IgM [131], ADA2-specific adenosine deaminase activity: blood & CD14+ monocytes. [73] [74] Absence of hypercoagulability. [131] Negative for antiphospholipid antibodies (APLAs). Some patients later develop a positive lupus anticoagulant (LA) level. Some have mild immunodeficiency. | |
| COPA Syndrome | Detailed info coming soon! | COPA | autosomal dominant | ||||||||||||||||
| Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis (PAAND) | Pyrin. Full details coming soon! | exon 2 MEFV, p.E244K | |||||||||||||||||
| OTULIN-related autoinflammatory syndrome (ORAS); Otulipenia; Autoinflammation, panniculitis, and dermatosis syndrome (AIPDS) | Details coming soon! | OTULIN | autosomal recessive | ||||||||||||||||
| Cleavage-resistant RIPK1-induced autoinflammatory) syndrome (CRIA ); Autoinflammatory with episodic fever and lymphadenopathy (AIEFL); Receptor Interacting Protein Kinase 1 (RIPK1)-Associated Immunodeficiency and Autoinflammation | Detailed info coming soon! | RIPK1 | autosomal dominant | ||||||||||||||||
| Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) | Details coming soon! | WDR1 | autosomal recessive | ||||||||||||||||
| POMP-related autoinflammation and immune dysregulation disease (PRAID); Proteasome-associated autoinflammatory syndrome-2 (PRAAS2). | Proteasome-associated. Details coming soon! | POMP | autosomal dominant | ||||||||||||||||
| NLRP1-associated autoinflammation with arthritis and dyskeratosis (NAIAD); Autoinflammation with arthritis and dyskeratosis (AIADK) | Details coming soon! | NLRP1 | autosomal recessive | ||||||||||||||||
| Pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa (PAPASH) | PSTPIP1-associated. Hidradenitis suppurativa associated. Details coming soon! | PSTPIP1 | |||||||||||||||||
| Pyoderma gangrenosum, acne and suppurative hidradenitis (PASH) Syndrome | PSTPIP1-associated. Hidradenitis suppurativa associated. Details coming soon! | PSTPIP1 | |||||||||||||||||
| IL-18–mediated PAP and recurrent MAS, IL-18–mediated pulmonary alveolar proteinosis (PAP) and recurrent Macrophage Activation Syndrome (MAS) | Interferonopathies, Il-18, MAS-associated | ||||||||||||||||||
| SAMD9L-associated autoinflammatory disease (SAMD9L-SAAD) | Interferonopaties | frameshift mutations in SAMD9L | |||||||||||||||||
| VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome | UBA1 (myeloid lineage-restricted somatic mutations in UBA1 ) | ||||||||||||||||||
| Pyoderma Gangrenosum, Acne and suppurative hidradenitis and ankylosing spondylitis (PASS) | PSTPIP1-associated. Hidradenitis suppurativa associated.Details coming soon! | PSTPIP1 | |||||||||||||||||
| Psoriatic Arthritis, pyoderma gangrenosum, acne and hidradenitis suppurativa (PsAPASH) | Hidradenitis suppurativa associated. Details coming soon! | unknown at this time | |||||||||||||||||
| Trisomy 8-associated autoinflammatory disease (TRIAD) | More details coming soon! | Trisomy 8 | |||||||||||||||||
| RELA-associated Familial Behçet’s-like Mucocutaneous Ulceration Syndrome (unofficial name) | More details coming soon! | RELA (truncation) | |||||||||||||||||
| HOIL-1 deficiency; immunodeficiency, autoinflammation, and amylopectinosis of cardiac and/or skeletal muscle | LUBAC deficiencies (HOIL-1 loss of function), Linear ubiquitin chain assembly complex (LUBAC), ubiqutin, NF-kB pathway | RBCK1 (HOIL-1 ) | autosomal recessive | ||||||||||||||||
| HOIP immunodeficiency, autoinflammation, some cases with amylopectinosis of cardiac and/or skeletal muscle and/or systemic lymphangiectasia | LUBAC deficiencies (HOIP loss of function), Linear ubiquitin chain assembly complex (LUBAC), ubiqutin, NF-kB pathway | RNF31 | recessive | ||||||||||||||||
| Pyoderma gangrenosum, Acne and ulcerative Colitis (PAC) | PSTPIP1-associated. Details coming soon! | PSTPIP1 | |||||||||||||||||
| Neonatal inflammatory skin and bowel disease (ADAM17) | ADAM17 | Autosomal Recessive | neonatal onset-infancy | ||||||||||||||||
| Retinal dystrophy-optic nerve edema-splenomegaly-anhidrosis-migraine headache syndrome; Optic nerve edema-splenomegaly syndrome; ROSAH syndrome | ALPK1 | Autosomal dominant | childhood | ||||||||||||||||
| AP1S3 | |||||||||||||||||||
| Cold-induced urticarial autoinflammatory syndrome related to factor XII activation | F12 | Autosomal Dominant | |||||||||||||||||
| Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK) syndrome | POMP-Related Systemic Autoinflammatory Diseases, Proteasome-associated, Autoinflammatory keratinization disease (AiKD) | homozygous single-nucleotide deletion in the 5′ UTR of POMP | Autosomal Recessive | ||||||||||||||||
| NEONATAL-ONSET CYTOPENIA WITH AUTOINFLAMMATION, RASH, AND HEMOPHAGOCYTOSIS (NOCARH) | CDC42-associated autoinflammatory diseases | CDC42 | Autosomal Dominant | infancy | Yes-see article links | See article links: Il-1 and Il-18 of note | |||||||||||||
| NEMO deleted exon 5–autoinflammatory syndrome | interferonopathies | novel splice variants in IKBKG |






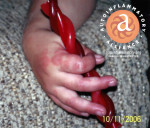















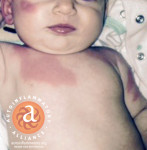





Main authors:
Karen Durrant RN, BSN–President of The Autoinflammatory Alliance (autoinflammatory.org), & Dr Juan Ignacio Aróstegui MD–Immunologist at the CDB Hospital Clínic in Barcelona, Spain & Director of La Unidad de Enfermedades Autoinflamatorias (autoinflamatorias.com)
Acknowledgements: A special thanks to the many medical doctors who have helped to make voluntary suggestions in regards to the original comparative chart, and suggestions for our new Autoinflammatory Search Database: Dr Juan Ignacio Aróstegui, Dr Hal Hoffman, Dr Raphaela Goldbach-Mansky, Dr Scott Canna, Dr Anna Simon, Dr Polly Ferguson, Dr Rebecca Marsh, Dr Daniel Kastner, Dr Luca Cantarini, Dr Véronique Hentgen, Dr Nico M. Wulffraat, Dr Kieron Leslie, Dr Lori Broderick, Dr Mikail Kostik, Dr Beata Wolska, Dr Joost Frenkel, Dr Dan Kastner, Dr Helen Lachmann, Dr Jonathan Hausmann, Dr Phillip Kahn, Dr Israel Andrews, and to all that have been using our materials to educate others about autoinflammatory diseases worldwide.
Thank you to Black Peacock SE, especially David Schwieler, Tommy Westerberg and Lotti Ungerth Fastmarken for all your amazing work on this database. Also to Nathan Durrant and Jennifer Tousseau for all your help on this, the original chart, and many projects. Our deepest thanks to all of The Autoinflammatory Alliance Board of Directors, & to all the patients & families who have supplied images for this chart, & support for the Autoinflammatory Alliance. You are our greatest inspiration and strength!
Great thanks to all of the doctors from the International Society of Systemic Auto-Inflammatory Diseases (ISSAID) for their research & dedication to patients with autoinflammatory diseases, plus the opportunity to present the original chart in a poster session at the Autoinflammation 2013 Congress. Thanks for the inspiration for this chart also go to: The Translational Autoinflammatory Disease Section at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) & The National Human Genome Research Institute at the National Institutes of Health (NIH); The Spanish Society of Pediatric Rheumatology (SERPE) & La Unidad de Enfermedades Autoinflamatorias; The French Centre de Référence des Maladies Auto-inflammatoires (CeRéMAI) & Le Club Rhumatismes et Inflamations; Dermatology Online Atlas. (DermIS Dermatology Information System), the Pediatric Rheumatology European Society (PReS),European League Against Rheumatism(EULAR), PRINTO, the EUROFEVER Project, the SHARE Consortium, The National Amyloidosis Centre, UK, The Interuniversity autoinflammation workgroup & Nijmegen center for Immunodeficiency and Autoinflammation (NCIA) of the Radboud Nijmegen University Medical Center, Nijmegen, The Netherlands, The American College of Rheumatology (ACR), CARRA, FAVOR & the many other research centers and doctors around the world.
Disclosure: All of the doctors involved in the authorship, review, editing and creation of this chart voluntarily donated their help for this educational reference, & received no financial compensation. Karen Durrant, RN has only received reimbursement only for for out-of-pocket travel costs from SOBI to attend a few meetings as a patient representative, but has received no personal financial compensation from any pharmaceutical company.
Swedish Orphan Biovitrum AB (Sobi), of Stockholm, Sweden provided the Autoinflammatory Alliance with an unrestricted grant in 2014 to support the development of this Autoinflammatory Search Database to help to educate medical professionals, and a future collection of disease-specific websites for patients. This grant, along with donated funds from the general public and patient community has helped to make this monumental project a reality, and we are so thankful!
Novartis Pharmaceuticals Canada Inc. provided The NOMID Alliance (now known as the Autoinflammatory Alliance) with an unrestricted grant in 2012 to help with the initial development & printing costs for the first comparative chart in print. An unrestricted grant from Swedish Orphan Biovitrum AB (Sobi) in 2013 supported many projects, including: the printing of the final comparative chart that we distributed at the 2013 ACR meeting, and 2014 PRYSM meeting, in addition to mailings to doctors worldwide. The NOMID Alliance has received a number of unrestricted grants at various times from Regeneron, Novartis & Sobi for grant-specific projects.
List of abbreviations:
- ACE: Angiotensin-converting enzyme (lab test)
- ADA2: Adenosine deaminase 2
- ANCA+ Vasculitis: Granulomatosis w/polyangiitis (GPA); Wegener’s
- ARDS: Acute Respiratory Distress Syndrome
- CD14+ monocytes: Cluster of differentiation 14 positive monocytes
- CD19: B-lymphocyte antigen CD19-aka Cluster of Differentiation 19
- CD25: Soluble interleukin-2-receptor
- CNS: Central Nervous System (involving the brain, spinal cord)
- CRP: C-reactive protein (lab test);
- DIC: Disseminated intravascular coagulation
- ESR: Erythrocyte sedimentation rate (lab test); Westergren ESR
- GI: Gastrointestinal (organs in the abdomen)
- HSP: Henoch–Schönlein purpura, anaphylactoid purpura
- ICP: Intracranial pressure
- IDDM: Insulin-Dependent Diabetes Mellitus
- IL-18: Interleukin 18
- LFTs: Liver function tests (lab test): AST, ALT, GGT, ALK Phos, Bilirubin
- NK cells: Natural killer cells
- PMNs: Polymorphonuclear leukocytes (on lab tests w/ WBC count)
- PTT: Partial thromboplastin time (lab test)
- SAA: Serum amyloid A protein (lab test)
- TSH: Thyroid-stimulating hormone (lab test); thyrotropin
- WBC: White Blood Count (lab test)